Ask any Indian Patent Attorney/Agent, are discoveries patentable in India? Straight comes the answer – No. The main argument for exclusion will be that patents are only granted for inventions which involve human intervention leading to inventive output not for identifying something which is inherent or already existing in nature. As India is new entrant to patenting phenomenon, whatever which is propagated by practicing patent attorneys/professionals is been conceived by others to follow. But I do not stand by it. Discoveries (not all) are patentable which is clearly evident from section 3. Section 3 of the Patents Act, 1970 lists what are not considered as patentable inventions. Section 3 has used three important words to support my contention, namely, 1) discovery; 2) mere discovery; and 3) invention. Explicit use of discovery, mere discovery and invention shows the intention of legislature that these words have different meaning and their patentability differs case by case. In section 3 (a), 3 (b) and 3 (p) the word ‘invention’ in been used whereas section 3 (c) and 3 (d) uses the word ‘discovery’ and ‘mere discovery’. Invention means technical outcome which results from or involve human intervention, for example, shaving razor, telephone etc. whereas discovery means something which is already there in nature but not known to public, for example, discovery of new elements of periodic table.
Is there a hidden catch?
Between discovery and mere discovery there lies a Silver lining. What stands between in and out is the word ‘mere’. In section 3 (c) both ‘discovery’ and ‘mere discovery’ are used which as follows –
“3 (c) the mere discovery of a scientific principle or the formulation of an abstract theory or discovery of any living thing or non-living substances occurring in nature”
According to section 3 (c), a scientific principle will not be considered as patentable invention if it is the mere discovery, emphasis is evidently there on the word ‘mere’. For example, in recently decided Calgon Carbon Corporation v. The Corporation of the City of North Bay, the Federal Court of Appeal, overturning the judgment of the motions judge, confirmed the patentability of a newly discovered use of an old method, so long as the new use has a practical application. The motion judge earlier found that the use of an old invention to prevent the crypto replication was a “mere discovery” and not a new invention and hence invalidated Calgon patent. While agreeing with the motions judge that a “mere discovery”, such a scientific observation, is not an invention, the court of Appeal distinguish “mere discoveries” from “inventions” under the Canadian Patent Act. The Court of Appeal explained that a discovery that UV light prevents crypto from replicating is a scientific observation for which no patent may be obtained since a scientific observation, on its own, has no practical application. However, the Court of Appeal held that finding a new, useful application for an existing method is more than a scientific observation. In Calgon, the patent claims are for what the Court of Appeal called “a practical solution to a practical problem”, which is more than a mere discovery.
Section 3 (c) further states, discovery of any living or non-living substances occurring in nature will not be considered as patentable invention. Here provision is straight enough that discovery of any living or non-living substances, whether or not involving technical ingenuity, will be not be patentable. Considering section 3 (c) alone, it can be construed that legislature have undoubtedly bifurcated ‘discovery’ and ‘mere discovery’ as well as their patentability issues. This argument can be made clearer and stronger by going through section 3 (d) which as follows –
“3 (d) the mere discovery of a new form of a known substance which does not result in the enhancement of the known efficacy of that substance of the mere discovery of any new property or new use for a known substance or of the mere use of a known process, machine or apparatus unless such known process results in a new product or employs at least one new reactant.
Explanation. – For the purposes of this clause, salts, esters, ethers, polymorphs, metabolites, pure form, particle size, isomers, mixtures of isomers, complexes, combinations and other derivatives of known substance shall be considered to be same substance, unless they differ significantly in properties with regard to efficacy;”
According to section 3 (c) mere discovery of a new form of known substance such as salts, esters, ethers, polymorphs, metabolites, pure form, particle size, isomers, mixtures of isomers, complexes, combinations and other derivatives of known substance will be patentable if the new form (product) results in the enhancement of the known efficacy of that substance. This means that the mere discovery of a new form with improved efficacy will be enough to overcome patentability bar. For example, if a new polymorphic form is having substantially improved thermal stability as compared to earlier known efficacy then the stated improved thermal stability will be enough to stand the requirement of inventive-step under section 2 (1) (ja), and thereby overcome the barrier of section 3 (d).
However, if a new form does not result in the enhancement of known efficacy then it will be not considered as patentable invention. The main argument for the exclusion will be that the mere discovery of a new form does not involve any technical merit to meet statutory bar of non-obviousness. For example, obtaining specific enantiomeric form by resolution of racemic mixture is the mere discovery as it already exists there, inherently. But section 3 (d) clearly connotes that mere discovery of a new form is patentable subject of meeting statutory condition, that its, enhancement of efficacy in respect of known efficacy. Thus, section 3 (d) diminishes the fate of trivial inventions but at the same time keeps the thumb up for well merit inventions.
Let Courts to decide
In absence of pertinent judicial decisions, my contention is still open for challenge. But explicit use of words ‘discovery’, ‘mere discovery’, and ‘invention’ cannot be ignored. Maybe after going through this, you may think again, are discoveries patentable in India?
Wednesday, March 29, 2006
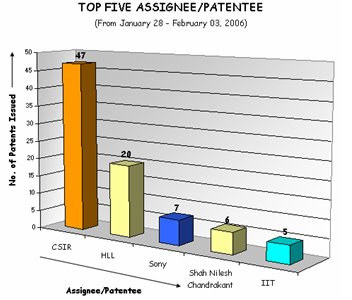
Top Five Assignees/Patentees (Jan. 28 - Feb. 03, 2006)
Indian Patent office publish list of patents issued every Friday. Here is provided top five assignees/patentees from January 28, 2006 – February 03, 2006.
An overview of Hatch-Waxman Act
Pre-Hatch-Waxman Regulatory Environment
In 1962, Kefauver-Harris Amendments to the Federal Food, Drug, and Cosmetic Act added a proof-of-efficacy requirement to new drug approvals. Before that time, the FDA approved drugs on safety parameters only. As a result all drug products approved between 1938 and 1962 (for safety only) were reviewed through the Drug Efficacy Study Implementation program to evaluate efficacy. Even today all brand-name companies are required to prove that new drugs are safe and effective for FDA approval. To prove this, brand-name companies are required to conduct human clinical trials and submit the results to the FDA with their New Drug Application (NDA). Those seeking to market a generic version of a post-1962 brand-name drug also had to perform their own safety and efficacy studies, much like the brand name companies. Hence due to the high costs involved few generic companies were interested in launching products in the US. As a result by 1984, the FDA estimated that there were approximately 150 brand-name drugs whose patents had expired for which there was no generic equivalent available.
Another factor complicating generic drug approval concerned the timing of when generic companies could perform their clinical tests. Even if a generic manufacturer gets access to the innovator clinical data making copies of a pharmaceutical product is not simple. Sourcing active ingredients, performing bio-equivalence studies, assuring quality, putting together a dossier, establishing patient information leaflets and going through the regulatory process can take 2-3 years. Manufacturing adds on another 3-6 months. Before Hatch-Waxman was enacted, a generic company could not begin the required FDA approval
process until the patents on the relevant brand-name product had expired. Consequently, patent protection was extended by 2-3½ years beyond the intended period.
Thus, at that time, FDA’s generic approval process coupled with the patent law, in effect, discouraged generic entry and extended the term of the brand-name company’s patent protection.
Not only the generic companies but also the brand-name pharmaceutical companies faced problems. The discovery and development of new drug is expensive and time consuming. To spur this investment, as well as to recoup investments made, brand name companies obtain patent protection to, prior to FDA approval of the drug product, to exclude others from making, using, or selling an invention for a number of years. Thus, the effective terms of many patents were shortened due to the time required for the FDA to ensure the safety and efficacy of the brand-name company’s drug product.
The Hatch-Waxman Act:
Congress passed the Hatch-Waxman Act to address the following issues:
- Cut generic approval costs: Rather than requiring a generic manufacturer to repeat the costly and time consuming NDA process, the act permitted the companies to file an Abbreviated New Drug Application (“ANDA”). Thereby allowing the generic applicants to rely on the brand-name company’s trade secret data to demonstrate the safety and efficacy of their products.
- Early-Experimental-Use-Doctrine: In the 1984 case of Roche Products v. Bolar Pharmaceutical Company, the US Court of Appeals for the Federal Circuit, held that a generic drug manufacturer's use of a drug under patent to develop information needed for market authorization was an act of patent infringement. This resulted in a pseudo patent extension for the innovator, since ANDA applicants had to wait until the patent expired prior to commencing any drug development activities. The so-called "Bolar Amendment,” embodied in 35USC §271(e) (1) of the patent law, and passed as part of the Hatch-Waxman Act, reversed the court's decision in the Roche v Bolar case and provided that “[I]t shall not be an act of infringement to make, use, offer to sell, or sell within the United States or import into the United States a patented invention...solely for uses reasonably related to the development and submission of information [to support a market approval to the FDA].” This provision allows generic manufacturers sufficient lead time to develop, perform necessary testing, and to seek US regulatory approval so they can be ready to launch their products upon expiration of a listed patent covering the innovator product.
- Compensate brand-name companies for the patent time lost in obtaining FDA approvals. Brand name companies were now allowed to obtain patent term extension as a measure of compensation of the patent time lost due to long regulatory approval process. The Act also introduced the concept of Market Exclusivity in the Federal Food, Drugs and Cosmetic Act. This was done to provide innovators with a minimum period of monopoly and protection against generic entry.
Thus the Hatch-Waxman Act balanced an expedited FDA approval process to speed generic entry with patent term extension and Market Exclusivity to ensure incentive for continues innovation. The Drug Price Competition and Patent Term Restoration Act of 1984 (Hatch-Waxman Amendments) added two new provisions to the Federal Food, Drug, and Cosmetic Act. Section 505(b)(2) : Hybrid New Drug Application (Changes to an Approved Drug)Section 505(j) : Abbreviated New Drug Application (Copy of Approved Drugs)
Abbreviated New Drug Application, 505(j) Filing
What is a Generic Drug? As per the U.S FDA, a generic drug product is the same as a brand name or a Reference Listed Drug (RLD) product, in dosage, safety, strength, how it is taken, quality, performance, and intended use. By law, a generic drug product must contain the identical amounts of the same active ingredient(s) as the brand name product. Before approving a generic drug product, FDA requires many rigorous tests and procedures to assure that the generic drug can be substituted for the brand name drug.
RLD (Reference Listed Drug): A Reference Listed Drug is an approved drug product to which generic versions are compared to show that they are bioequivalent. All RLD’s are listed in the FDA's list of Approved Drug Products with Therapeutic Equivalence Evaluations also known as the Orange Book. A drug company seeking approval to market a generic equivalent must refer to the Reference Listed Drug in its Abbreviated New Drug Application (ANDA). By designating a single reference listed drug as the standard to which all generic versions must be shown to be bioequivalent, FDA hopes to avoid possible significant variations among generic drugs and their brand name counterpart.
Definition of an Abbreviated New Drug Application (ANDA): It contains data that, when submitted to FDA's Center for Drug Evaluation and Research, Office of Generic Drugs, provides for the review and ultimate approval of a generic drug product. Generic drug applications are called "abbreviated" because they are generally not required to include preclinical (animal) and clinical (human) data to establish safety and effectiveness. Instead, a generic applicant must only scientifically demonstrate that its product is bioequivalent (i.e., performs in the same manner as the innovator drug). Once approved, an applicant may manufacture and market the generic drug product to provide a safe, effective and a low cost alternative to the public. At the time of filing an ANDA the applicant must certify the clause under which he wishes to file the application and hence issue a certificate in this regard: Para I filing : This certification is made when the ANDA applicant is aware that there may be a patent in force that may cover the RLD, but that the NDA holder has for whatever reason decided not to list the patent with the agency. If an ANDA applicant makes a Paragraph I certification, the FDA may approve its application immediately, as long as the FDA determines that the ANDA otherwise meets the approval requirements; or Para II filing : A Paragraph II certification is appropriate when there is a patent listed in the Orange Book, but it has expired. The ANDA applicant simply certifies that the patent has expired, and provides the patent number and the date the patent expired. An ANDA application containing such a certification is eligible for immediate effective approval if the ANDA applicant has otherwise met the FDA approval requirements; or Para III filing : A Paragraph III certification acknowledges that there is a listed patent on the RLD that has not expired and that the ANDA applicant does not plan to market its product prior to the expiration of the patent. In this instance, the law precludes the FDA from approving the application until the patent has expired; or
Para IV filing : An application containing a Paragraph IV certification signifies that the ANDA applicant plans to challenge one or more of the listed patents. The ANDA holder hence claims that the patent is invalid, unenforceable, or will not be infringed by the manufacture, use, or sale of the generic product. A generic applicant makes a Paragraph IV certification only when its intent is to market the drug product prior to the expiration date of the patent.
This article is submitted by Ashish Mohan, Dept. of Pharmaceutical Management, NIPER, Mohali, India (ashish_k001@yahoo.co.in)
Tuesday, March 28, 2006
Patentable Inventions: Part II
Method of Treatment
Section 3 (i) states, any process for the medicinal, surgical, curative, prophylactic, diagnostic, therapeutic or other treatment of human beings or any process for a similar treatment of animals to render them free of disease or to increase their economic value or that of their products is not patentable. As the main argument for the exclusion the definition of “industrial applicability” is used. Patent may however be obtained for surgical, therapeutic or diagnostic instrument or apparatus.
A method of treatment of malignant tumor cells and method of removal of dental plaque and carries are not patentable as per section 3 (i) but an application of substance to human body purely for cosmetic purpose does not fall within the scope of this section. Prophylactic treatment such as vaccination, inoculation is not patentable. For example, prophylactic immuno-therapy in animals is regarded as therapy where the term therapy includes prevention as well as treatment or cure of diseases. In Joos vs. Commissioner of Patent (1973) RFC 59, it was held that to be treatment in relevant senses it seems that the purpose of the application to the body whether a substance or a process must be the arrest or cure of a disease or diseased condition or correcting of some malfunction or amelioration of some incapacity or disability. It was held in Lee Pharmaceuticals application (1978) RFC 51 that since one of the results of grant pits and fissures in teeth was to prevent the onset of dental decay, the purpose of the treatment was therapeutic rather than cosmetics.
In Oral Health Products Inc. (Habtead’s Application, (1977) RFC 612), the claims to a method of removing dental plaque and/or caries were refused, as was a claim to a method of cleaning teeth, which embraced both curative and cosmetic effects. This decision has been followed in ICI Lid’s Application (not reported), where a claim was refused to a method of cleaning teeth which removed both plaque and stains; it was argued that when applied to perfectly healthy teeth the method was purely cosmetic, but the hearing officer observed that practically all medicinal treatments which are preventive in nature (such as vaccination) must at times be applied to people who would have remained healthy anyway, but they remained medicinal treatments.
Surgery is defined as the treatment of disease or injury by operation or manipulation. It is not limited to cutting the body but includes manipulation such as the setting of broken bones or relocating dislocated joints, and also dental surgery. In general, any operation on the body which required the skill and knowledge of a surgeon would be regarded as surgery. For example, in Unilever limited (Davis) Application, [1983] RFC 219, Falconer J observed that any method of surgical treatment, whether curative, prophylactic or cosmetic, is not patentable. This view, which was obiter, was cited by the hearing officer in Occidental Petroleum Corporation’s Application (not reported) in refusing to allow claims to a method of implanting an embryo transplant from a donor mammal into the uterus of a recipient mammal, since the method would necessarily have to be carried out by a surgeon or veterinary surgeon.
Section 3 (i) is however limited to methods practiced on the human or animal body excluding plants. Also methods of diagnosis performed on tissues or fluids, which have been permanently removed from the body, are outside the scope of section 3 (i). To further clarify this section “body” means a living body, therefore, a method practiced on a dead body, for example a process that determines the cause of death, fall outside the scope of this section. Hence, diagnostic methods are patentable if they are used outside of living human or animal bodies i.e. body tissues or fluids like urine or blood.
Saturday, March 25, 2006
List of APIs going Off-patent in 2006
Here is provided the list of important APIs going off-patent in 2006 with their latest updates.
Simvastatin: (Expiring on June 23, 2006)
U.S. Patent No. 4,444,784 (Hoffman Patent) titled “Antihypercholesterolemic compounds” discloses and claims Simvastatin and use thereof for treating hypercholesterolemia. The application for Hoffman Patent was filed on December 18, 1980 by Merck & Co. and issued on April 24, 1984. The term of Hoffman Patent will be 17 years calculated from the date of issuance of patent.
Hoffman Patent was eligible for patent term extension under 35 USC § 156 (Hatch-Waxman Act) and was granted extension of 1704 days, extending the patent validity period from April 24, 2001 to December 23, 2005. Later Hoffman Patent also received additional six months pediatric exclusivity, yet again extending the patent validity period till June 23, 2006.
Earlier Ranbaxy and Ivax filed Para IV certification challenging the validity and enforceability of two orange book listed patents – US RE36481 and US RE36520 – expiring in 2008 and 2009 respectively. Ranbaxy being the first to file Para IV certification was entitled for 180-days exclusivity period. In response, Merck de-listed both the US patents and thereby preventing Ranbaxy from getting 180-days exclusivity period. Recently, Merck has granted Dr. Reddy’s Laboratories exclusive rights to market generic version of Zocor (Simvastatin) as authorized generics once Simvastatin go off-patent.
Pravastatin: (Expiring on April 20, 2006)
U.S. Patent No. 4,346,227 (Terahara Patent) titled “ML-236B Derivatives and their preparation” discloses and claims pravastatin. The application for Terahara Patent was filed on June 05, 1981 by Sankyo Company Limited and issued on August 24, 1982. The term of Terahara Patent will be 20 years calculated from the date of US filing.
Terahara Patent was eligible for patent term extension under 35 USC § 156 and was granted extension of 1598 days, extending the patent validity period from June 05, 2001 to October 20, 2005. Later Terahara Patent also received additional six months pediatric exclusivity, yet again extending the patent validity period till April 20, 2006.
Earlier Teva filed ANDA seeking permission to sell generic pravastatin once Terahara Patent expires in April 2006 (Para III) and assertion that generic version would not violate three patents on the drug (Para IV certification). Teva being the first-to-file Para IV certification is entitled for 180-days exclusivity after favorable ruling from US District Court for the District of Columbia involving complex tussle with US FDA.
Sertraline Hydrochloride: (Expiring on June 30, 2006)
U.S. Patent No. 4,536,518 (Welch Patent) titled “Antidepressant derivatives of cis-4-phenyl-1, 2, 3, 4-tetrahydro-1-naphthaleamine” discloses and claims Sertraline hydrochloride and use thereof for combating mental depression in a mentally-depressed patient. The application for Welch Patent was filed on November 01, 1979 by Pfizer and issued on August 20, 1985. The term of Welch Patent will be 17 years calculated from the date of issuance.
Welch Patent was eligible for patent term extension under 35 USC §156 and was granted extension of 1228 days, extending the patent validity period from August 20, 2002 to December 30, 2005. Later Terahara Patent also received additional six months pediatric exclusivity, yet again extending the patent validity period till June 30, 2006.
Earlier, Ivax filed ANDA seeking permission to sell generic sertraline HCl once Welch Patent expires in June 2006 (Para III) and assertion that generic version would not violate US Patent No. 5,248,699 (listed with orange book) on the drug (Para IV certification). Pfizer sued Ivax for patent infringement and latter settled their dispute out of the court. Ivax is, however, entitled for 180-days exclusivity once Welch Patent goes off-patent.
Finasteride: (Expiring on June 19, 2006)
U.S. Patent No. 4,760,071 (Rasmusson Patent) titled “17b-N-monosubstituted carbamoyl-4-aza-5a-androst-1-en-3-ones which are active as testosterone 5 a-reductase inhibitor” discloses and claims finasteride and use thereof for treating the hyperandrogenic condition of acne vulgaris, seborrhea, femal hirsutism, and benign prostatic hypertrophy and for inhibiting testosterone 5a-reductase. The application for Rasmusson Patent was filed on November 21, 1985 by Merck & Co. and issued on July 26, 1988. The term of Rasmusson Patent will be 17 years calculated from the date of issuance.
Rasmusson Patent was eligible for patent term extension under 35 USC §156 and was granted extension of 328 days, extending the patent validity period from July 26, 2005 to June 19, 2006.
Earlier, Merck sued Dr. Reddy’s in response of Para IV certification filed with US FDA. Later Merck and Dr. Reddy’s settled patent dispute and Merck granted Dr. Reddy’s the exclusive right to market generic version of Proscar (Finasteride) after patent expiry as authorized generic.
Esomeprazole: (Expiring on October 19, 2006)
U.S. Patent No. 4,738,974 (Brandstrom Patent) titled “Base addition salts of Omeprazole” discloses and claims novel salts of the Omeprazole and use thereof for inhibiting gastric acid secretion and for treating gastrointestinal inflammatory diseases or cytoprotective effects. The application for Brandstrom Patent was filed on April 21, 1986 by Aktiebolaget Hassle and issued on April 19, 1988. The term of Brandstorm Patent will be 17 years calculated from the date of issuance.
Brandstorm Patent was eligible for patent term extension under 35 USC §156 and was granted extension of 1 year, extending the patent validity period from April 19, 2005 to April 19, 2006. Later Brandstrom Patent also received additional six months pediatric exclusivity, yet again extending the patent validity period till October 19, 2006.
Earlier, AstraZeneca sued Ivax, Teva and Ranbaxy for patent infringement in the U.S. District Court for the District of New Jersey in response of Para IV certification filed with US FDA seeking permission to market generic versions of Nexium (Esomperazole).
Desloratadine: (Expiring on October 21, 2006)
U.S. Patent No. 4,659,716 (Villani Patent) titled “Antihistaminic 8-(halo)-substituted 6, 11-dihydro-11-(4-piperidylidene)-5H-benzo [5, 6] cyclohepta [1, 2-b] pyridines” discloses and claims desloratadine and pharmaceutical compositions thereof. Villani Patent also claims method of treating allergic reactions using desloratadine. The application for Villani Patent was filed on March 12, 1986 and issued on April 21, 1987. The term of Villani Patent will be 17 years calculated from the date of issuance.
Villani Patent was eligible for patent term extension under 35 USC §156 and was granted extension of 2 year, extending the patent validity period from April 21, 2004 to April 21, 2006. Later Villani Patent also received additional six months pediatric exclusivity, yet again extending the patent validity period till October 21, 2006.
According to US FDA official website, there is no Para IV certification filed for desloratadine.
Terbinafine: (Expiring on December 30, 2006)
U.S. Patent No. 4,755,534 (Stuetz Patent) titled “Propenylamines, pharmaceutical compositions containing them and their use as pharmaceuticals” discloses and claims terbinafine and use thereof for treating diseases or infections caused by mycetes. The application for Stuetz Patent was filed on September 04, 1984 by Sandoz Ltd. and issued on July 05, 1988. The term of Stuetz Patent will be 17 years calculated from the date of issuance
Stuetz Patent was eligible for patent term extension under 35 USC §156 and was granted extension of 543 days, extending the patent validity period from July 05, 2005 to December 30, 2006.
Earlier, Novartis sued Dr. Reddy’s for patent infringement in response of Para IV certification filed with USFDA but later patent challenge was withdrawn by Dr. Reddy’s.
Zolpidem Tartrate: (Expiring on October 21, 2006)
U.S. Patent No. 4,382,938 (Kaplan Patent) titled “Imidazo [1,2-a] pyridine derivatives and their application as pharmaceuticals” discloses and claims zolpidem tartrate and use thereof for providing anxiolytic effect. The application for Kaplan Patent was filed on October 21, 1981 by Synthelabo and issued on May 10, 1983. The term of Kaplan Patent will be 20 years calculated from the date of US filing.
Kaplan Patent was eligible for patent term extension under 35 USC §156 and was granted extension of 5 years, extending the patent validity period from October 21, 2001 to October 21, 2006.
Balsalazide Disodium: (Expiring on July 08, 2006)
U.S. Patent No. 4,412,992 (Chan Patent) titled “2-hydroxy-5-phenylazobenzoic acid derivatives and method of treating ulcerative colitis therewith” discloses and claims balsalazide disodium and use thereof for treating ulcerative colitis. The application for Chan Patent was filed on July 08, 1981 by Biorex Laboratories Limited and issued on November 01, 1983. The term of Chan Patent will be 20 years calculated from the date of US filing.
Chan Patent was eligible for patent term extension under 35 USC §156 and was granted extension of 5 years, extending the patent validity period from July 08, 2001 to July 08, 2006.
According to US FDA official website, there is no Para IV certification filed for balsalazide disodium.
Nedocromil Sodium: (Expiring on October 02, 2006)
U.S. Patent No. 4,474,787 (Cairns Patent) titled “7,6 Dioxo-4H, 6H-pyrano [3,2-g] quinoline dicarboxylic acids and anti-allergic use thereof” discloses and claims Nedocromil sodium and use thereof for treating a condition involving an antibody antigen reaction or a reflex pathway. The application for Cairns Patent was filed on February 02, 1982 by Fisons Limited and issued on October 02, 1984. The term of Cairns Patent will be 17 years calculated from the date of issuance.
Cairns Patent eligible for patent term extension under 35 USC §156 and was granted extension of 5 years, extending the patent validity period from October 02, 2001 to October 02, 2006.
Perindopril Erbumine: (Expiring on August 21, 2006)
U.S. Patent No. 4,508,729 (Vincent Patent) titled “Substituted iminodiacids, their preparation and pharmaceutical compositions containing them” discloses and claims perindopril erbumine and use thereof for treating hypertension. The application for Vincent Patent was filed on October 02, 1981 by Adir and issued on April 02, 1985. The term of Vincent Patent will be 17 years calculated from the date of issuance of patent.
Vincent Patent eligible for patent term extension under 35 USC §156 and was granted extension of 1601 days, extending the patent validity period from April 02, 2002 to August 21, 2006.
According to US FDA official website, there is no Para IV certification filed for perindopril erbumine.
Lomefloxacin HCl: (Expired on February 21, 2006)
U.S. Patent No. 4,528,287 (Itoh Patent) titled “6-fluro-1, 4-dihydroxy-4-oxo-7-substituted piperazinylquinoline-3-carboxylic acids and method for preparing the same” discloses and claims Lomefloxacin HCl and use thereof for inhibiting bacteria and for increasing urinary excretion. The application for Itoh Patent was filed on September 17, 1984 by Hokuriku Pharmaceuticals Co. Ltd. and issued on July 09, 1985. The term of Itoh Patent will be 20 years calculated from the date of US filing.
Itoh Patent eligible for patent term extension under 35 USC §156 and was granted extension of 522 days, extending the patent validity period from September 17, 2004 to February 21, 2006.
Fulvestrant: (Expiring on October 01, 2006)
U.S. Patent No. 4,659,516 (Bowler Patent) titled “Steroid derivatives” discloses and claims fulvestrant and use thereof for producing an antiestrogenic effect. The application for Bowler Patent was filed on October 01, 1984 by Imperial Chemicals Industries Ltd. and issued on April 21, 1987. The term of Bowler Patent will be 20 years calculated from the date of US filing.
Bowler Patent eligible for patent term extension under 35 USC §156 and was granted extension of 2 year, extending the patent validity period from October 01, 2004 to October 01, 2006.
According to US FDA official website, there is no Para IV certification filed for fulvestrant.
Mivacurium Chloride: (Expired on January 22, 2006)
U.S. Patent No. 4,761,418 (Swaringen Patent) titled “Novel Compounds” discloses and claims Mivacurium chloride and use thereof for producing muscle relaxation. The application for Swaringen Patent was filed on July 17, 1985 by Burroughs Wellcome Co. and issued on August 02, 1988. The term of Swaringen Patent will be 17 years calculated from date of issuance.
Swaringen Patent eligible for patent term extension under 35 USC §156 and was granted extension of 173 days, extending the patent validity period from August 02, 2005 to January 22, 2006.
Thursday, March 23, 2006
Patentable inventions: Part I
Any invention falling within the scope of section 3 of the Patents Act, 1970 is not considered to be a patentable invention. Section 3, particularly section 3 (d), always attracted great debate among the patent professionals/attorneys. Being a statutory provision, section 3 is always open for judicial interpretation but in absence of case laws clear explanation is still lacking. Unlike US computer software, business method, and method of treatment are not patentable in India. Till date, section 3 has been amended twice to broaden the limiting scope and to ensure that only well merit inventions see the light of patent protection. Let us go through section 3 and scrutinize what is in and what is out.
Frivolous or Against Public Policy
Section 3(a) states, an invention which is frivolous or which claims anything obviously contrary to well established natural laws is not patentable. In patent law, any invention to be patentable must have industrial utility and must be capable of industrial application. For example, in 2005 UK Patent Office rejected a patent application titled “Energy lasting increasing endlessly (ELIE)” considering that the disclosed invention was related to a perpetual machine and was thus not capable of industrial application. Examiner argued that disclosed invention alleged to operate in a manner which was clearly contrary to well-established physical laws and thus not patentable. Section 3 (b) further states, an invention the primary or intended use of commercial exploitation of which could be contrary public order or morality or which claims serious prejudice to human, animal or plant life or health or to the environment is not patentable. For example, a surgical device for performing abortion may be consider as non-patentable in India on ground on public morality. This provision may differ country to country because what is consider immoral in India, may not be considered immoral in US, for example, gambling devices. Inventions intended to perform fraudulent activities are considered to be non-patentable, for example, burglary.
Method of Agriculture or horticulture
Section 3 (h) states, a method of agriculture or horticulture is not patentable. Unlike in US, inventions related to cultivation of plants are not patentable in India. For example, a method of produce a new form of a known plant which involves modifying the conditions under which natural phenomena would purse their inevitable course is not patentable. Indian Patent Application claiming a method of producing mushroom plant (445/Del/93) and a method of cultivation of an algae (264/Del/79) was rejected by the Patent Office considering that the production of mushrooms and cultivation of an algae is analogues to agriculture because they belong to plant kingdom and therefore fall within the scope of section 3 (h). Inventions such as ploughing or sowing equipments, machines used for cultivation, and devices for storing agricultural or horticultural products are patentable. However, cultivation of micro-organisms and silk rearing do not fall within the scope of section 3 (h).
Monday, March 20, 2006
Generic Tamiflu: What if Patent is granted?
Indian pharmaceutical companies has already commenced to manufacture and market generic versions of Tamiflu (Oseltamivir Phosphate), the patent application for which is still pending for grant with Delhi Patent Office. Indian Patent Application No. 396/DEL/96 (“396 patent application”) covering avian flu drug Tamiflu was filed under section 5 (2) (“mail-box” application) for which examination has to be initiated after 1.1.2005. Currently 396 patent application is subject to pre-grant opposition under section 25 (1) of the Patents Act, 1970 which is likely to be taken up by Patent Office in near future. But the important question that arises here is what will be the fate of pharmaceutical companies that are manufacturing and marketing generic version of Tamiflu if patent is granted?
According to section 11A of the Patents Act, 1970 all patent applications will be publish after completion of 18 months, after which application will be open for pre-grant opposition. Further section 11A (7) states that an applicant, on and from the date of publication of the application for patent and until the date of grant of a patent in respect of such application, will have the like privileges and rights as if a patent for the invention had been granted on the date of publication of the application with a proviso that
the applicant will not be entitled to institute any proceedings for infringement until the patent has been granted with a further proviso that
the rights of a patentee in respect of applications made under section 5 (2) before the 1st day of January, 2005 shall accrue from the date of grant of the patent with yet another proviso that
after a patent is granted in respect of applications made under 5 (2), the patent-holder will only be entitled to receive reasonable royalty from such enterprises which have made significant investment and were producing and marketing the concerned product prior to the 1.1.2005 and which continue to manufacture the product covered by the patent on the date of grant of the patent and no infringement proceedings will be instituted against such enterprises.
Prior to January 01, 2005
Proviso of section 11A (7) of the Patents Act, 1970 clearly states that a patent-holder cannot a bring an infringement suit against generic manufactures which have made significant investment and were producing and marketing the concerned product prior to the 1.1.2005 and will continue to manufacture the product covered by the patent on the date of grant of the patent by paying reasonable royalty to the patentee.
Is this section provides Tamiflu generics a shield against infringement action?
Answer is No.
Proviso clearly emphasizes that generic manufactures should be producing and marketing the patented product prior to 1.1.2005, the requirement which is not met in the case of Tamiflu. Generic manufactures, however, got market approval from Drug Controller General of India (DCGI) in 2006 and started marketing their generic versions from February 2006. This clearly shows that in case of patent issuance, Roche/Gilead is not bond by the proviso of section 11A (7) and can institute patent infringement lawsuit against generic manufactures.
Generic Dilemma
Generic Tamiflu is already having presence in the Indian market which has recently witness the introduction of product patent in pharmaceutical sector. Considering that Gilead/Roche has successfully procured Tamiflu patent worldwide, the likelihood of obtaining patent in India cannot be ruled out. Tamiflu, however, do not fall under the scope of proviso of section 11 A (7). It is the generics that will be in dilemma if patent is issued. Two critical issues that will certainly cause problem to generics: 1) timely vacating their generic version from market, and 2) Stockpiling.
IIT Kharagpur to commence three-year LLB programme in IPR
India’s premier institute IIT Kharagpur is all set to kick-start its law school with a three-year LLB programmes in intellectual property rights (IPR) and one-year post-graduate diploma programme for practicing professionals and corporates from the next academic session. IIT Kharagpur has also signed a technical collaboration agreement with George Washington University for exchange of faculty and students and draft of curriculum through its School of Law.
Saturday, March 18, 2006
Ciba Specialty filed Patent Lawsuit against RiTEK
Ciba Specialty Chemicals has filed a lawsuit for patent infringement in Germany and Netherlands against RiTEK Corporation and its European subsidiaries. The lawsuit is filed in response of Ciba’s patented CD-R dye technology alleging that RiTEK import and sell RiTEK-manufactured CD-R which infringes Ciba’s patents.
Thursday, March 16, 2006
Generics are back in Game
Judge Rodney Sippel of the U.S. District Court for the eastern district of Missouri, on January 17, 2006, has ruled under summary judgment in favor of the generic challengers, clearing their way to launch generic versions of Toprol-XL in U.S. market. This is, however, a pyrrhic victory for generic manufactures after successive para IV defeats in Lipitor (Atorvastatin Calcium, Ranbaxy losing to Pfizer), Accupril (Quinapril Hydrochloride, Ranbaxy losing to Pfizer), Norvasc (Amlodipine Besylate, Dr. Reddy’s loses to Pfizer) and Zyprexa (Olanzapine, Dr. Reddy’s loses Eli Lilly). The court has favored suits filed by KV Pharmaceutical Co, Andrx Corp. and Eon Labs Inc. (a unit of Novartis AG), challenging the validity and enforceability of Orange Book listed patents. Astra, which earlier contended before the District Court that its patents on Toprol-XL will survive their patent term, has made an appeal against the judgment in the U.S. Court of Appeals maintaining that both US patents (5001161 and 5081154) are valid and enforceable.
Targeting Para IV
To recap, in February 2004, AstraZeneca filed a patent infringement lawsuit against Andrx Corp. in the United States District Court for the District of Delaware as a result of a Paragraph IV certification letter submitted by Andrx concerning its intent to manufacture and sell generic form of Toprol-XL in the 50 mg dose form in US. Again, in July 2004 AstraZeneca filed a second patent infringement against Andrx Corp. in the same District Court following the Andrx’s notification that it had filed ANDA under paragraph IV with USFDA for seeking approval to market generic form of Toprol-XL 50 mg dose form. Astra advocated that Andrx infringes its patents covering Toprol-XL. In the same year, AstraZeneca also filed a lawsuit in the District Court for the District of Delaware against generic manufacture, Eon Labs, for patent infringement following the Eon’s move for market approval of generic form of Toprol-XL for all dose forms under paragraph IV. Moreover, later in the same year, AstraZeneca filed legal proceedings against KV Pharmaceutical Co. in the District Court for the Eastern District of Missouri this time following the KV’s notification regarding ANDA submission under paragraph IV for market approval of generic form of Toprol-XL 50 mg dose form. Earlier to this AstraZeneca filed a suit in May 2003 against KV Pharmaceutical Co. for patent infringement by the 200mg dose form of Toprol-XL and again in August 2003 for patent infringement by the 100 mg dose form. Pre-trial Discovery All of these pending patent litigations later consolidated for pre-trial discovery purposes and motion practice in the U.S. District Court for the Eastern District of Missouri, which do not include a jury. A pre-trial discovery means that a party to a lawsuit should not be surprised by its adversary on the day of trial, and therefore has the right to full discovery, prior to trial, of all relevant evidence, including documents (broadly defined to include virtually every tangible and intangible form of record keeping) and witness testimony related to the matter at issue in the litigation. In other words, each party to the suit attempts to “discover” relevant or pertinent facts. However, in this trial consideration was of the validity and enforceability of two AstraZeneca patents (US Patent No. 5,081,154 and 5,001,161) relating to active ingredient (metoprolol succinate per se) and composition of Toprol-XL (extended release tablet of metoprolol succinate) and to determine whether the defendants in the case infringe those patents. Both ‘154 and ‘161 patent were due to expire in September of 2007.
Targeting Para IV
To recap, in February 2004, AstraZeneca filed a patent infringement lawsuit against Andrx Corp. in the United States District Court for the District of Delaware as a result of a Paragraph IV certification letter submitted by Andrx concerning its intent to manufacture and sell generic form of Toprol-XL in the 50 mg dose form in US. Again, in July 2004 AstraZeneca filed a second patent infringement against Andrx Corp. in the same District Court following the Andrx’s notification that it had filed ANDA under paragraph IV with USFDA for seeking approval to market generic form of Toprol-XL 50 mg dose form. Astra advocated that Andrx infringes its patents covering Toprol-XL. In the same year, AstraZeneca also filed a lawsuit in the District Court for the District of Delaware against generic manufacture, Eon Labs, for patent infringement following the Eon’s move for market approval of generic form of Toprol-XL for all dose forms under paragraph IV. Moreover, later in the same year, AstraZeneca filed legal proceedings against KV Pharmaceutical Co. in the District Court for the Eastern District of Missouri this time following the KV’s notification regarding ANDA submission under paragraph IV for market approval of generic form of Toprol-XL 50 mg dose form. Earlier to this AstraZeneca filed a suit in May 2003 against KV Pharmaceutical Co. for patent infringement by the 200mg dose form of Toprol-XL and again in August 2003 for patent infringement by the 100 mg dose form. Pre-trial Discovery All of these pending patent litigations later consolidated for pre-trial discovery purposes and motion practice in the U.S. District Court for the Eastern District of Missouri, which do not include a jury. A pre-trial discovery means that a party to a lawsuit should not be surprised by its adversary on the day of trial, and therefore has the right to full discovery, prior to trial, of all relevant evidence, including documents (broadly defined to include virtually every tangible and intangible form of record keeping) and witness testimony related to the matter at issue in the litigation. In other words, each party to the suit attempts to “discover” relevant or pertinent facts. However, in this trial consideration was of the validity and enforceability of two AstraZeneca patents (US Patent No. 5,081,154 and 5,001,161) relating to active ingredient (metoprolol succinate per se) and composition of Toprol-XL (extended release tablet of metoprolol succinate) and to determine whether the defendants in the case infringe those patents. Both ‘154 and ‘161 patent were due to expire in September of 2007.
Back in Game!
The judgment day belongs to generics, a much needed victory for all Para IV seekers. Judge Rodney Sippel in his summary judgment found that AstraZeneca’s patents (US Patent No. 5,081,154 and 5,001,161) on Toprol-XL were “both invalid due to double patenting and are unenforceable due to inequitable conduct.” Astra’s both ‘161 patent and ‘154 patent are invalidated on the basis of double patenting over the US 4,780,318 patent and are considered unenforceable based on Astra’s inequitable conduct in the prosecution of these patents in the United States Patent & Trademark Office concerning inventorship dispute over metoprolol succinate with their competitor named Lejus Medical. However, it was also argued during proceedings that Astra intentionally did not name the correct inventors in Astra’s prosecution of patents in suit. Court found that Astra’s failure was done with an intent to deceive the patent examiner about the inventorship dispute and to avoid questions concerning Astra’s ability to claim priority to the ‘318 patent application and to avoid potential prior art concerning metoprolol succinate.
This means that now generic challengers can launch their generic copies of Toprol-XL in US. Andrx being the first to file ANDA under Para IV will be, however, entitled to 180-days exclusivity on the 50mg strength of generic Toprol-XL under Hatch-Waxman Act. But Andrx’s approval and launch of generic Toprol-XL will depend on FDA clearance and approval requirements and outcome of subsequent legal proceeding made by AstraZeneca in US Court of Appeals. Even though, Astra has made an appeal against the judgment in the U.S. Court of Appeals, what cannot be denied is that Generics challengers are back in game.
Tuesday, March 14, 2006
Abstracting Patent Specification
A brief abstract of the technical disclosure made in the specification usually commences on a separate sheet, preferably following the claims, under the heading “Abstract” or “Abstract of the Disclosure.” The abstract as required under section 10 (4) (d) of the Patents Act, 1970 may not exceed 150 words in length (Rule 13 (7) (c), the Patents Rules, 2003). Abstracting is done to constitute an efficient instrument for the purpose of searching in the particular technical field, in particular to enable Patent Office and the public generally to determine quickly from a cursory inspection the nature and gist of the technical disclosure. The content of a patent abstract should include that which is new in the art to which the invention pertains.
Content of a Patent Abstract
A patent abstract is a concise summary of the technical disclosure made in the specification and should clearly indicate the technical field to which the invention belongs, technical problem to which the invention relates and the solution to the problem through the invention and principle use or uses of the invention (Rule 13 (7) (b), the Patents Rules, 2003). For example, if the specification discloses the nature of an improvement in old apparatus, process, product, or composition, the abstract should include the technical disclosure of the improvement. However, if the specification relates to modifications or alternatives, the abstract should mention by way of example the preferred modification or alternative. In certain patent specifications, particularly those for compounds or compositions, wherein the process for making and/or the use thereof are not obvious, the abstract should set forth a process for making and/or a use thereof. Where necessary, the abstract should contain the chemical formula, which characterizes the invention. If necessary most relevant figure of the drawings should also be included in the abstract.
Where applicable, the abstract should include the following –
if a machine or apparatus, its organization and operation;
if a chemical compound, its identity and use;
if a composition, its ingredients;
if a process, the steps.
Is it necessary?
A patent abstract is a statutory requirement which forms an integral part of complete disclosure of the invention. The preparation of the patent abstract is the responsibility of the applicant under duty of disclosure. However, the Controller may amend the abstract during prosecution for providing better information to third parties under section 10 (4) (d) (i) of the Patents Act, 1970.
Come full circle: Wining ‘Pioglitazone’ Battle
Takeda has come full circle by wining a Para IV patent litigation and successfully defending the validity and enforceability of their orange book listed US Patent No. 4,687,777 covering the active ingredient of their oral anti-diabetic drug “ACTOS”, that is, pioglitazone hydrochloride. On February 21, 2006 Judge Denis Cote of the U.S. District Court for the Southern District of New York, has ruled in the favor of Takeda maintaining that ‘777 patent is valid and enforceable, and thereby preventing Food and Drug Administration (FDA) from approving the abbreviated new drug applications (ANDAs) filed by Alphapharm and Mylan. This victory is outcome of weak arguments made by the generic movers during the court proceedings, miserably failing to meet their homework.
Para IV: Tempting Generic Move
On July 15, 2003 Watson Pharmaceutical, Alphapharm, Ranbaxy, and Mylan all filed ANDAs seeking approval to market 15mg, 30mg, and 45mg pioglitazone hydrochloride tablets. Ranbaxy and Watson filed Para III certifications with respect to the ‘777 patent, not challenging the validity of ‘777 patent but both filed Para IV certifications relating to the composition claims in the combination use patents. Alphapharm and Mylan filed a Para IV certifications with respect of the ‘777 patent, claiming that it is invalid. In their Para IV certifications, both Alphapharm and Mylan claimed that the ’777 patent was invalid due to obviousness and forwarded notice of Para IV certification to Takeda that included detailed statements under 21 USC 21§ 355 (j) (2) (b) (ii) clarifying the legal basis of their positions. Mylan in their notice described that '777 patent was invalid in the light of Compound 16 of U.S Patent No. 4,287,200 ('200 patent). Mylan also asserted that Compound 16 of '200 patent is identical to a Compound 14 identified in a prior publication of Dr. Sohda. Alphapharm also contended in their notice that pioglitazone covered by '777 patent is prima facie obvious over the prior art compound, disclosed in '200 patent and published article of Dr. Sohda.
But by the time of trial, each defendant completely altered their approach to the invalidity issue expressed in their statement. Mylan abandoned its obviousness issue, and pursued an inequitable conduct claim at trial whereas Alphapharm altered its obviousness argument in several substantial ways. The trial was based to resolve the challenges made by the defendants to the validity and enforceability of Takeda's 777 patent which took place from Jan. 17 to Jan 30 2006.
Missing the line of concrete ‘obvious’ evidence
Submission of Alphapharm's Para IV certification statement recognized that during the trial proceeding they have the burden to prove by clear and convincing evidence that the prior art provides some reason or motivation for a person of ordinary skill to make pioglitazone. To substantiate its contention, Alphapharm, identified only two sources of prior art: '200 patent and prior publication of Dr. Sohda. But during the trial proceedings they also argued about the divisional patents to the '200 patent and a few articles in scientific journals. Referring to '200 patent, Alphapharm argued that the patent generically disclosed pioglitazone, and identified three compounds related to pioglitazone: compounds 16, 40, and 42, emphasizing particularly on compound 42 which is methyl homolog of the ethyl compounds covered by Claim 1 of the '777 patent, arguing that an exchange of an ethyl group for a methyl group would have been obvious. Coming to Dr. Sodha's article, Alphapharm argued that discussion referring to compound 58 would lead one of ordinary skill in the art to choose compound 58 as the lead compound for further development. However, this argument was not part of their Para IV certification statement. Alphapharm argued that the prior art clearly identified compound 58 as a lead compound warranting further investigation, and that the application of a few, obvious chemical processes would have produce pioglitazone. Alphapharm also argued various technical flaws made in '777 patent. Court, however, found that Alphapharm's arguments fails to show any persuasive evidence, much less by clear and convincing evidence, that one with ordinary skill in the art have had any reasonable expectation or motivation based on the prior art for synthesizing pioglitazone.
No Inequitable Conduct
During the trial proceeding, Mylan contended that the ‘777 patent is invalid because Takeda engaged in inequitable conduct, claiming that Takeda intentionally made material misrepresentations in its presentation made in the ‘777 patent when applying to the Patent and Trademark Office in 1986. However, this argument was not part of their Para IV certification statement. Mylan also raised few other issues to attack Takeda in its presentation made in the ‘777 patent. Mylan argues that Takeda fails to disclose to USPTO a compound already disclosed in the prior art, specifically in publication of Dr. Sohda, which is parent structure for the left hand moiety of the pioglitazone molecule. Mylan argues that if Takeda had been revealed that prior known compound along with its comparable activity and toxicity to pioglitazone then USPTO "might" have concluded that pioglitazone is not patentable over prior art. But Mylan in their contention failed to prove that Takeda has made any misstatements or has failed to provide the USPTO material information. The Court of law found that Takeda has clearly identified the relevant prior art including both Dr. Sohda and '200 patent. Judge Dennis Cote in his 124-page ruling wrote, “Mylan has failed to carry its burden of showing either a material misstatement or omission. It has also failed to present any evidence regarding intent to deceive the Patent and Trademark Office."
Still in the Game
Valid and enforceable ‘777 patent has shifted the tempting generic move back to innovator Takeda, keeping their faith intact with its patents. The Court’s ruling now prevents generic manufacturers from selling and marketing pioglitazone tablets until the '777 patent expires in 2011. Other U.S. patents covering certain methods of treatment using pioglitazone and certain compositions that include pioglitazone will expire in 2016. In another development, Pfizer has successfully defended Lipitor in Finland, after losing in lower court to Ranbaxy. The Helsinki Court of Appeal has granted a preliminary injunction against Ranbaxy, barring Ranbaxy from selling and marketing generic version of lipitor. These unfavorable rulings will definitely not affect the Para IV strategy of generic challengers but to make the game they need to restructure it, not technically but in line of Para IV jurisprudential precedents.
Sunday, March 12, 2006
Shire sued yet another Generic Challenger, Teva, for infringing Adderall XR patents
Shire Laboratories had sued Teva Pharmaceuticals for alleged infringement of two of Shire’s US Patent Nos. 6,322,819 and 6,605,300 covering attention deficit hyperactivity disorder drug - Adderall XR. Both 819’ and 300’ patents are due to expire on October 21, 2018 and April 21, 2019 with pediatric exclusivity.
The lawsuit had been filed in the U.S District Court for the Eastern District of Pennsylvania as a result of an abbreviated new drug application (ANDA) filed by Teva with Food and Drug Administration for the marketing approval of generic versions of Adderall XR.
Earlier on 24th February 2003, Shire Laboratories sued Barr for infringement of U.S. Patent No. 6,322,819 in the U.S. District Court for the Southern District of New York in response of an abbreviated new drug application (ANDA) submitted by Barr with Food and Drug Administration (FDA) seeking approval to market generic Adderall XR in the United States. The ‘819 patent was only orange book listed patent at the time of Barr ANDA submission in November 2002. Later, Shire submitted one more U.S. Patent No. 6,605,300 with FDA to be listed with orange book and in response of that Barr subsequently amended its ANDA to include a certification to the ‘300 patent and provided Shire with notice of that amendment. On September 02, 2003 Shire filed second lawsuit against Barr for infringement of ‘300 patent in the U.S District Court for the Southern District of New York in response of amended ANDA submission.
In December 2003 Shires also sued Impax for infringement of ‘819 and ‘300 patents in the U.S. District Court for the Southern District of New York in response of an ANDA filed by Impax in November 2003 with FDA seeking approval to market generic Adderall XR of 30mg strength prior to the expiration dates of the ‘819 and ‘300 patents in the United States. In January 2005, Shires filed second lawsuit against Impax for infringement of ‘819 and ‘300 patents in response of an ANDA submission seeking approval to market generic Adderall XR of 5mg, 10mg, 15mg, 20mg and 25mg strengths.
In December 2004, Shire even after receiving notification that Colony Pharmaceuticals has submitted an ANDA with FDA seeking approval to market generic Adderall XR of 5mg, 10mg, 15mg, 20mg, 25mg and 30mg strengths prior to the expiration dates of the ‘819 and ‘300 patents, decided not to sue it.
In December 2004, Shire even after receiving notification that Colony Pharmaceuticals has submitted an ANDA with FDA seeking approval to market generic Adderall XR of 5mg, 10mg, 15mg, 20mg, 25mg and 30mg strengths prior to the expiration dates of the ‘819 and ‘300 patents, decided not to sue it.
On October 20, 2005 Shires announced that it has filed yet another lawsuit against Barr and Impax for infringement of U.S. Patent No. 6,913,768 in the U.S District Court of the Southern District of New York in response of an ANDAs filed by Barr and Impax. The ‘768 patent was issued to Shire on July 05, 2005. This case, however, does not effect the ongoing litigations in the Southern District of New York between Shire and Barr involving ‘819 and ‘300 patents which later consolidated in December 2003 and scheduled to go to trial in January 2006. In January 2006, Shire and Impax settled all pending patent litigation concerning Adderall XR under an agreement that Impax will be permitted to market generic Adderall XR in the United States no later than January 01, 2010 and will pay Shire a royalty from those sales. In certain conditions, such as launch of another generic version of Adderall XR, Impax may be permitted to enter the market as Shire’s authorized generic. No payments to Impax are involved in the settlement agreement.
Friday, March 10, 2006
Settling Unfinished Patent Infringement Disputes
Allergan and Alcon have reached an agreement to settle two patent infringement lawsuits initiated by Allergan. Under the terms of agreement, Allergan will dismiss both patent suits with prejudice, and also grant Alcon a co-exclusive license with Allergan for brimonidine tartrate 0.15% ophthalmic solution. The settlement grants Alcon the right to introduce its brimonidine 0.15% product on September 30, 2009 or earlier if certain market conditions occur, the primary condition being a trigger based generally on the extent to which prescriptions of Alphagan P 0.15% have been converted to other brimonidine-containing products marketed by Allergan. In return, Alcon will pay royalties to Allergan on sales of the product once it has been launched.
Allergan filed the first lawsuit in the U.S District Court for the District of Delaware contending that Alcon’s proposed brimonidine 0.15% product infringes Allergan’s orange book listed US Patent Nos. 6,673,337 and 6,641,834. Both ‘337 and ‘834 patent are valid up to July 2021 and January 2022 (with pediatric exclusivity). The lawsuit was in response to a New Drug Application (NDA) filed by Alcon under section 505(b) (2) of the Federal Food, Drug and Cosmetic Act seeking approval for marketing generic brimonidine tartrate ophthalmic solution 0.15% product in the United States. Alcon’s product received tentative Food and Drug Administration (FDA) approval on 28th February 2005, but final approval was withheld pending resolution of the litigation.
The second lawsuit was filed in the U.S. District Court for the District of California alleging infringement of U.S Patent Nos. 6,166,012 and 6,492,361 directed to self-preserved antibiotic products. Alcon believes this lawsuit was intended to target its leading ophthalmic ocular antibiotic Vigamox (0.5% moxifloxacin HCl ophthalmic solution).
Allergan filed the first lawsuit in the U.S District Court for the District of Delaware contending that Alcon’s proposed brimonidine 0.15% product infringes Allergan’s orange book listed US Patent Nos. 6,673,337 and 6,641,834. Both ‘337 and ‘834 patent are valid up to July 2021 and January 2022 (with pediatric exclusivity). The lawsuit was in response to a New Drug Application (NDA) filed by Alcon under section 505(b) (2) of the Federal Food, Drug and Cosmetic Act seeking approval for marketing generic brimonidine tartrate ophthalmic solution 0.15% product in the United States. Alcon’s product received tentative Food and Drug Administration (FDA) approval on 28th February 2005, but final approval was withheld pending resolution of the litigation.
The second lawsuit was filed in the U.S. District Court for the District of California alleging infringement of U.S Patent Nos. 6,166,012 and 6,492,361 directed to self-preserved antibiotic products. Alcon believes this lawsuit was intended to target its leading ophthalmic ocular antibiotic Vigamox (0.5% moxifloxacin HCl ophthalmic solution).
Thursday, March 09, 2006
AstraZeneca sues Ivax for infringing Nexium
AstraZeneca is back again, this time again defending its heartburn drug Nexium against generic challenger, Ivax. On March 08, 2006 AstraZeneca filed a patent lawsuit in the U.S. District Court for the District of New Jersey against IVAX and IVAX’s parent, Teva Pharmaceuticals Industries Ltd., for willing infringement of AstraZeneca’s patents protecting Nexium, generically known esomeprazole magnesium.
The lawsuit was filed in response of an abbreviated new drug application (ANDA) filed under Para IV certification by Ivax with Food and Drug Administration (FDA) seeking approval for generic version of Nexium in the US prior to the expiration of the five AstraZeneca orange book listed patents: 5,714,504; 5,877,192; 6,369,085; 6,428,810; and 6,875,872. The expiration dates for these patents range from 2014 through to 2019.
Earlier in November 2005, AstraZeneca also sued Ranbaxy for willfully infringing AstraZeneca’s patents protecting Nexium and filed a lawsuit in response of an ANDA submission for esomperazole magnesium delayed-release capsules under Para IV certification with Food and Drug Administration (FDA). The lawsuit was filed in the same District Court.
The lawsuit was filed in response of an abbreviated new drug application (ANDA) filed under Para IV certification by Ivax with Food and Drug Administration (FDA) seeking approval for generic version of Nexium in the US prior to the expiration of the five AstraZeneca orange book listed patents: 5,714,504; 5,877,192; 6,369,085; 6,428,810; and 6,875,872. The expiration dates for these patents range from 2014 through to 2019.
Earlier in November 2005, AstraZeneca also sued Ranbaxy for willfully infringing AstraZeneca’s patents protecting Nexium and filed a lawsuit in response of an ANDA submission for esomperazole magnesium delayed-release capsules under Para IV certification with Food and Drug Administration (FDA). The lawsuit was filed in the same District Court.
Wednesday, March 08, 2006
US Court of Appeals stays injunction on Eligard Drug
US Court of Appeals has stayed an injunction against the QLT barring it from manufacturing or selling prostate cancer drug Eligard, generically known as leuprolide acetate, in the US. The injunction has been stayed pending the Court’s decision on whether to grant a permanent stay of the injunction.
On February 27, 2006 the U.S. District Court for the Northern District of Illinois Eastern Division granted an injunction enjoining QLT, Sanofi-Aventis and their subsidiaries from promoting, manufacturing, selling and offering for sale QLT USA’s Eligard product in the US until U.S. Patent No. 4,728,721 expires on May 1, 2006. The Court further ordered QLT and Sanofi-Synthelabo to recall any Eligard products that they still own and provide a voluntary recall program to allow physicians, wholesalers and distributors, who wish to do so, to return Eligard for a full refund. The Court also granted a stay of the injunction for seven days from the date of grant of injunction.
Earlier in 2003, TAP sued Atrix Laboratoires Inc. (now QLT USA) and Sanofi-Synthelabo alleging that QLT USA’s Eligard product infringes ‘721 patent. In response QLT and Sanofi-Synthelabo challenged the validity and enforceability of the ‘721 patent, which was later in December 2005 rejected by Judge James Zagel ruling that TAP ‘721 patent is valid.
Tuesday, March 07, 2006
Forest Laboratories receives patent term extension for Lexapro
US Patent No. RE34712 covering escitalopram oxalate has received a patent term extension under 35 USC § 156 for 828 days. Marketed by Forest laboratories under the brand name Lexapro was earlier due to expire on June 08, 2009 and December 08, 2009 with pediatric exclusivity, will now be valid upto September 14, 2011 and upto March 14, 2012 with pediatric exclusivity. This extension will be a major boost up for Forest Laboratories in its battle against generic competitors.
In 2004, Forest received notification from two generic manufactures, namely, Ivax Pharmaceuticals and Alphapharm Pty. Ltd., that they had filed abbreviated new drug applications (ANDAs) with a Para IV certification with the Food and Drug Administration (FDA) for approval of generic version of escitalopram oxalate. In response, Forest along with its licensing partner Lundbeck A/S filed a suit against Ivax jointly with Cipla Ltd. and Alphapharm. However, on 04th October ’05, Forest and Lundbeck entered into a Settlement Agreement with Alphapharm but does not settle the pending patent litigation against Ivax and Cipla. On October 26, 2005 the Federal District Court, District of Delaware, rescheduled the start of the trial from December 05, 2005 to March 15, 2006.
Roche obtain first pharmaceutical product patent in India
The Controller General of Patents has issued first pharmaceutical product patent no. 198952 (the ‘952) to Swiss drug maker F. Hoffman-La-Roche for its biotech hepatitis drug Pegasys under the new patent regime paving way to reintroduction of product patent in India. However, any interested party may still challenge the ‘952 patent under post-grant provision (u/s 25(2)) within a year of grant of patent.
Subscribe to:
Comments (Atom)